Well done to Richard on winning the medical student category of our ‘Unofficial Guide to Medicine Essay Competition’ for his essay on “Consider the development of personalised medicine. What are the benefits and drawbacks of selecting appropriate therapies based on the context of one’s genetic code?”

I am a 5th year MB/PhD student at the University of Cambridge, currently in my research phase at the Babraham Institute whilst continuing medical education. My current ambition is to become a clinician-scientist, taking the best of both worlds! I have a particular interest in the development of novel cancer therapeutics.
Consider the development of personalised medicine. What are the benefits and drawbacks of selecting appropriate therapies based on the context of one’s genetic code?
Personalised medicine in cancer: Progress at an almighty cost
During my undergraduate studies, the story of imatinib mesylate always struck me as the poster child of personalised medicine. Here was a drug that could inhibit the BCR-ABL protein, formed in a chromosome translocation event in the development of approximately 90% of chronic myeloid leukaemia1. Overall survival in the imatinib arm of phase III trials was an impressive 85%2, demonstrating an ability to specifically target cancers based on their mutational profile. Coupled with our lectures on high throughput genomic sequencing, we could be forgiven for believing that the age of personalised medicine was not starting, but had already been here for a long time. It was therefore, hugely disappointing when I entered the wards in my fourth year to see paclitaxel, carboplatin and other chemotherapies designed in the 70s and 80’s, which target rapidly dividing cells in an indiscriminate manner still forming the bulk of cancer treatments. Were my lecturers wrong to say that personalised medicine will be the way forward?
In an ever increasingly cash strapped National Health Service (NHS), the cost of medications is constantly being reviewed by the National Institute for Health and Care Excellence (NICE). NICE aims to achieve the most quality adjusted life years (QALY’s) for a given amount of money, following an adapted utilitarian argument. There have been many recent tragic news stories of patients denied modern cancer therapies they believed could extend their lives, for example AA Gill’s damning final article3. Ultimately, these therapies are falling short of NICE criteria, so much so that in 2010 the coalition government created the cancer drugs fund to circumnavigate the uncomfortable truth that many of these drugs are very poor value for money, preventing the NHS from widely utilising personalised therapy. But, is this simply a case of greedy pharmaceutical CEOs? As ever, it is more complex. Production costs for targeted therapies is high, extortionately high, but this is not so much due to the drug itself, rather the hundreds of drugs that showed initial promise but never made the final hurdle. In a 2012 study, a staggering 63.5% of cancer drugs that entered stage III clinical trials failed to progress4, and oncology has one of the lowest likelihood of approval for modern therapies5. How can this be?
The argument of poor clinical trial design is a popular explanation and has already been extensively commented on elsewhere4,5. However, other more basic arguments exist for a high rate of failure such as our lack of appropriate pre-clinical models. Cell cultures do not consider tumour microenvironment, immune interaction or nutrient supply to list a few examples. When it comes to animal models, nude mice xenografts have all the aforementioned disadvantages of cell culture. More complex arguments exist for transgenic mice, such as is the induced mutation occurring at the correct stage of the tumorigenesis process and would this affect the phenotypic effect? However, despite all the complex theoretical arguments against the basic models used there is a much more fundamental rotten core at the science of medicine from bench to bedside, and that is the irreproducibility of data. At the clinical level, this is beautifully illustrated in Ben Goldacre’s TED talk in 20116. However, it is just as prominent at the lab-bench level, with approximately only 20% of pre-clinical studies being reproducible7,8. It is simply unfathomable to think that 80% of the data we are using to design personalised medicines is flawed and yet this appears to be the picture that is emerging. It is therefore unsurprising that we develop medications with such high costs and with the majority failing to prove long-term efficacy. Together, these are two major roadblocks preventing NICE and clinical commissioning groups opting for the latest personalised therapies, when preventative measures such as smoking cessation and generic chemotherapies offer better cost/benefit outcomes.
One ever decreasing cost is our ability to evaluate the genetic code of tumours to enable appropriate therapeutic targeting. It is now possible to do whole genome sequencing for approximately $1000 (USD), which would have simply been unthinkable only a decade ago. Expensive narrow approaches such as Fluorescent In situ hybridisation (used for chromosomal translocations) are being phased out for the more comprehensive next generation sequencing technologies. Furthermore, the breadth of our genetic knowledge is ever expanding, with epigenetic modifications being evaluated by bisulphite sequencing as an example. Even as we discover the greater complexities of cancer, such as the heterogenicity of subclones, technologies such as detection of circulating tumour DNA are revolutionising our ability to select appropriate personalised medicines. Whilst we are not quite there, the sheer rate of development in genetic analysis is something to hold a great deal of hope for.
Unfortunately, even when effective, therapy resistance has proved the bane of almost every personalised targeted therapy we have tried. It is now infamously demonstrated with the images of a man who had extensive metastatic melanoma which underwent apparent complete regression under vemurafenib, a drug targeted to the V600E mutated BRAF gene present approximately 45% of melanomas9, only for the tumour to reappear 16 weeks later10. Tackling this resistance has invited a number of approaches, with some advocating further downstream targets such as transcription factors11, whilst others have suggested drug holidays since it appears that resistant tumours have a Darwinian growth disadvantage to their non-resistant parental tumour, meaning that the two clones can effectively undergo an evolutionary fight amongst themselves12. However, by far the most popular idea is that of combination therapies which target signalling pathways of mutated genes at multiple points and/or targets the exact repertoire of mutations that each individual patient’s tumour has13. However, even if successful there are potential pitfalls to be had in the translation of combinational/ poly- therapy.
In an age of evidence based medicine, personalised treatment presents a theoretical and statistical problem. Each patient’s genetic mutations are exactly that, individual to the patient. By definition they are a N-of-1 study for their particular cocktail of drugs. This is in stark contrast to the methods to date, where we try desperately hard to categorise in order to enable effective comparison in randomised control trials. How are we best to evaluate drugs intended for polytherapy, in a trial environment which to date has been so reliant upon monotherapy? Even when drug combinations are utilised today, they are the same combination across a large patient cohort to enable randomised analysis. This may mean our range of combinations may be limited to those in which we can recruit sufficient patients with the correct combination of mutations (diagrammatically represented in figure 1). However, there is an argument that current analysis is only a result of our own, sometimes arbitrary, categorisation of variables which may be wholly inappropriate, as argued by Dr Schork who feels that N-of-1 studies are to be championed in evidence based medicine as an improvement since it will appropriately take into account all the patients’ variables14. However, how statistical analysis could ever result from this is not clear. Furthermore, there is an ethical dilemma. Given the complications of the evidence base of a patient’s polytherapy and indeed the complexity of mechanism of action, we head rapidly into a time old problem but one which is vital to respect, that of patient autonomy. Since we cannot analyse how previous patients have responded to a personalised cocktail of drugs, it makes discussions on side effects, prognostication and appropriateness for palliation close to impossible. This challenge will place clinicians in the very uncomfortable position of simply not knowing any answers to many questions patients would wish to know about their therapeutic regime.
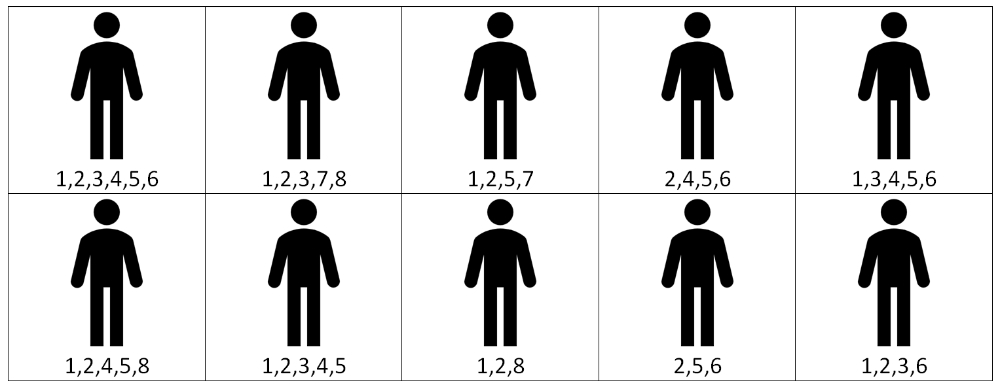 Figure 1: Diagramatic example of study design limitations. If theraputic drugs are avaliable for mutations 1 through to 4, a trial of drugs to 1 and 2 would encompass 70% of patients. However a trial of 1 to 4 would only encompass 20% of patients.
Figure 1: Diagramatic example of study design limitations. If theraputic drugs are avaliable for mutations 1 through to 4, a trial of drugs to 1 and 2 would encompass 70% of patients. However a trial of 1 to 4 would only encompass 20% of patients.
The idea of sending a patient’s blood, containing circulating DNA of their specific cancer so that a cocktail of therapies can be concocted, is both highly tantalising but currently not feasible. However, with examples like imatinib acting as a proof of principle, the benefits of personalised medicines in cancer are clear to see, even though Imatinib has encountered its own problems with resistance mechanisms15. Other therapies have also proven to be effective, albeit not curative, but we have become fat of our own success, complacent with the steady trickle of targeted therapies emerging. Even now cancers such as pancreatic cancer have a woeful 3% 5-year survival rate16, attributed not only to its difficult surgical location but also to its extreme heterogeneity. A change of tone is therefore needed, where we cannot be complacent with an 80% failure rate in our preclinical research alone, simply because we are at the cutting edge of science. So how can the medical profession counter these growing concerns with personalised medicines, from the ethical to the scientific? One solution is for the clinician to become more involved in the basic science that develops these therapies, not only to enable more knowledgeable communication with patients, but to aid in the preclinical design and development. Such ambitions are already promoted with an increasing trend towards clinical involvement in academic research, and the GMC placing the “doctor as scientist” as a fundamental pillar of medical education in the ‘tomorrow’s doctor’ framework. Only by unifying basic, translational and clinical arms of drug development, previously relatively dichotomous, can the enormous benefits of personalised medicine in cancer outweigh the significant disadvantages.
Bibliography
- Salesse, S. & Verfaillie, C. M. BCR/ABL: from molecular mechanisms of leukemia induction to treatment of chronic myelogenous leukemia. Oncogene 21, 8547–8559 (2002).
- Deininger, M. et al. International Randomized Study of Interferon Vs STI571 (IRIS) 8-Year Follow up: Sustained Survival and Low Risk for Progression or Events in Patients with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Treated with Imatinib. Blood 114, (2015).
- AA Gill: Final article describes cancer treatment – BBC News. Available at: http://www.bbc.co.uk/news/uk-38280057. (Accessed: 12th January 2017)
- Gan, H. K., You, B., Pond, G. R. & Chen, E. X. Assumptions of expected benefits in randomized phase III trials evaluating systemic treatments for cancer. J. Natl. Cancer Inst. 104, 590–8 (2012).
- Hay, M., Thomas, D. W., Craighead, J. L., Economides, C. & Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51 (2014).
- Ben Goldacre: Battling bad science | TED Talk | TED.com. Available at: https://www.ted.com/talks/ben_goldacre_battling_bad_science. (Accessed: 12th January 2017)
- Prinz, F., Schlange, T. & Asadullah, K. Believe it or not: how much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 10, 712–712 (2011).
- Begley, C. G. & Ellis, L. M. Drug development: Raise standards for preclinical cancer research. Nature 483, 531–533 (2012).
- Ascierto, P. A. et al. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 10, 85 (2012).
- Wagle, N. et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 29, 3085–96 (2011).
- Johnston, S. J. & Carroll, J. S. Transcription factors and chromatin proteins as therapeutic targets in cancer. Biochim. Biophys. Acta – Rev. Cancer 1855, 183–192 (2015).
- Sun, C. et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122 (2014).
- Caunt, C. J., Sale, M. J., Smith, P. D. & Cook, S. J. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat. Rev. Cancer 15, 577–592 (2015).
- Schork, N. J. Personalized medicine: Time for one-person trials. Nature 520, 609–611 (2015).
- Bitencourt, R., Zalcberg, I. & Louro, I. D. Imatinib resistance: a review of alternative inhibitors in chronic myeloid leukemia. Rev. Bras. Hematol. Hemoter. 33, 470–5 (2011).
- Pancreatic cancer statistics | Cancer Research UK. Available at: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer#heading-Two. (Accessed: 13th January 2017)
Feedback
Your essay on personalised medicine in cancer was a delight to read. You have taken a very original approach to this by focusing on one topic within the hugely expanding world of personalised medicine which has clearly enabled you to develop a very in-depth understanding of the subject. You have used relevant references throughout your essay to support your writing and have researched this topic in great detail but have also shown a lot of independent thought and have expressed many of your own opinions on the issues surrounding personalised medicine and the possible solutions. Excellent use of a figure to support your point on study design limitations. Well done, this is a very well written, concise, scientific essay. Richard’s essay on personalised medicine in cancer medicine was a truly fantastic read. He commanded a remarkable knowledge and understanding of the subject matter, which demonstrates an understanding of the literature that far beyond the context of this short essay. The presentation of these arguments was written eloquently in a logical matter. What helped to make this essay so outstanding was the detailed analysis of the problems surrounding the current paradigm of the research. All in all, this essay was excellent. Well done Richard.